Aplastische Anämie (AA)
1 Was ist eine AA?
2 Symptome
3 Diagnostik
4 Klinischer Verlauf
5 Therapie
6 Prognose
7 Register
1 Was ist eine AA?
1.1 Allgemeines
Die Aplastische Anämie ist eine nicht bösartige (nicht maligne) hämatologische Erkrankung. Ihr liegt eine Störung der Knochenmarkfunktion zugrunde, bei der es zu einer verminderten Bildung von Blutzellen kommt.
Je nach Ursache einer Aplastischen Anämie unterscheidet man angeborene (z.B. Diamond-Blackfan- oder Fanconi-Anämien, Telomeropathien) von erworbenen Formen. Beide Formen können in jedem Lebensalter auftreten.
Störungen der Blutbildung nach einer Chemotherapie oder Bestrahlung werden nicht als Aplastische Anämie bezeichnet.
1.2 Vorkommen (Epidemiologie)
Die Krankheitshäufigkeit (Inzidenz) liegt in Mitteleuropa bei 2-3 Neuerkrankungen pro 1 Million Menschen und Jahr. Damit gehört die Aplastische Anämie zu den sehr seltenen Erkrankungen. Die meisten Betroffenen erkranken zwischen dem 10. und 25. Lebensjahr sowie oberhalb des 60. Lebensjahres, wobei beide Geschlechter gleich häufig betroffen sind.
1.3 Entstehung (Pathogenese)
Studien zufolge greift bei der erworbenen Aplastischen Anämie ein Teil des eigenen Immunsystems Zellen im Knochenmark an. Es handelt sich dabei um eine Unterart der Lymphozyten, die durch diesen Autoimmunprozess die Bildung neuer Blutzellen verhindert. In einigen Fällen der erworbenen Aplastischen Anämie vermutet man Arznei- oder giftige (toxische) Stoffe (ca. 20 %) oder eine Virusinfektion (ca. 5 %) als Ursache.
In den meisten Fällen einer erworbenen Aplastischen Anämie (ca. 75 %) lässt sich eine Ursache für die Entstehung der Erkrankung jedoch nicht ermitteln, sodass der Ursprung der Erkrankung ungeklärt (idiopathisch) bleibt. Es gibt jedoch auch Fälle, bei denen eine angeborene Veränderung für das Auftreten oder den Verlauf einer Aplastischen Anämie verantwortlich ist.
Neuere Studien zeigen, dass ein relevanter Teil der Patienten mit anscheinend erworbener Aplastischer Anämie an einer angeborenen Form leidet, die sich erst spät klinisch zeigt. Aktuell liegt der Anteil der angeborenen Fälle, die erst im Erwachsenenalter erkannt werden, bei 5-15 % aller Aplastischen Anämien bei Erwachsenen. Es ist anzunehmen, dass dieser Anteil durch die besseren Untersuchungsmöglichkeiten weiter ansteigen wird. Relevant ist dies, da sich bei dieser Patientengruppe die Therapieempfehlung von den Fällen mit erworbener Aplastischer Anämie unterscheidet. Dies ist insbesondere in Hinblick auf eine Stammzelltransplantation von Bedeutung.
1.4 Diagnosekriterien und Klassifizierung
Damit man bei einer Erkrankung von einer Aplastischen Anämie ausgehen kann, müssen die folgenden Kriterien erfüllt sein:
- Die Anzahl der Zellen im Knochenmark (Zellularität) liegt bei weniger als 25 % gegenüber gesundem Knochenmark, wobei die Bewertung anhand einer Knochenmarkbiopsie erfolgt. Die Zellproduktion kann im Rahmen der Erkrankung vermindert sein (hypoplastisch) oder komplett fehlen (aplastisch).
- Verminderung von zwei (Bizytopenie) bzw. drei Zellreihen (Tri- bzw. Panzytopenie) in unterschiedlicher Ausprägung im Blutausstrich.
- Es gibt keinen Anhalt für eine Bindegewebs(neu-)bildung im Knochenmark (Fibrose) bzw. einen Knochenmarkbefall durch bösartige (maligne) oder knochenmarkfremde Zellen.
- Zusätzlich dürfen keine bedeutenden Zellveränderungen (Dysplasien) der Blutbildung (Hämatopoese) vorliegen.
- Es wurde kürzlich keine Strahlen- oder Chemotherapie durchgeführt, die eine Störung der Knochenmarkfunktion (Knochenmarkinsuffizienz) erklären könnte.
- Es fand kein Kontakt mit radioaktiver Strahlung statt.
Die Unterteilung der Aplastischen Anämie erfolgt anhand der Blutwerte (siehe nachfolgende Tabelle) in:
- nicht schwere Aplastische Anämie = nSAA („non-severe AA“)/mäßig schwere Aplastische Anämie („Moderate AA“)
- schwere Aplastische Anämie = SAA („severe AA“)
- sehr schwere Aplastische Anämie = vSAA („very severe AA“)
und ist von entscheidender Bedeutung für Prognose und Therapie.
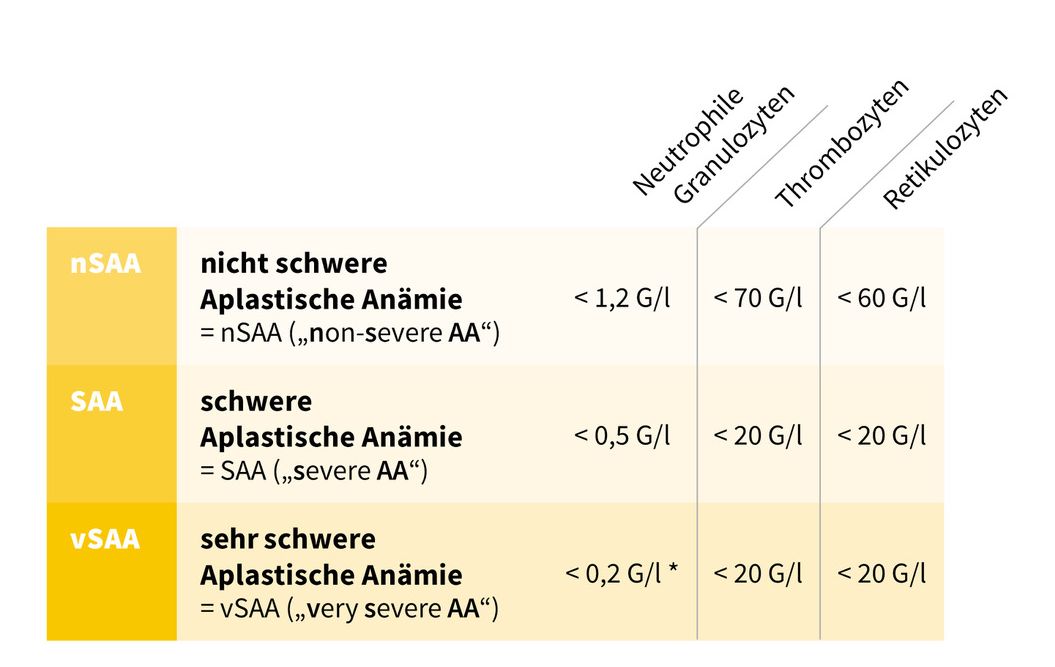
Klassifikation der Aplastischen Anämie anhand des Blutbilds (Zellzählung und -ausstrich). Zwei von drei Kriterien müssen erfüllt sein. *Für die Klassifikation als vSAA muss das Kriterium Granulozyten < 0,2 G/l zwingend erfüllt sein.
2 Symptome
2.1 Blutarmut
Eine Verminderung an Sauerstoff-transportierenden roten Blutzellen (Erythrozyten) kann insbesondere bei körperlicher Belastung Schwäche, Müdigkeit und Kurzatmigkeit bis hin zu Herzrasen hervorrufen. Außerdem fallen Patienten mit Anämie häufig durch Blässe insbesondere der Handinnenflächen auf, wobei das Vorhandensein einer Blässe nicht als Beweis einer Blutarmut zu verstehen ist.
2.2 Verstärkte Infektanfälligkeit
Durch eine verminderte Anzahl der weißen Blutkörperchen (Leukozyten) ist der Körper verstärkt infektgefährdet. Da das körpereigene Abwehrsystem bei einer erniedrigten Zahl an neutrophilen Granulozyten, einer Unterart der weißen Blutkörperchen, nicht ausreichend funktioniert, kann ein solcher Infekt innerhalb von Stunden einen lebensbedrohlichen Verlauf nehmen und zu einer Blutvergiftung führen.
Daher ist es wichtig, dass Sie bei Auftreten von Fieber sofort Ihren behandelnden Arzt informieren. Fieber ist definiert als eine Körpertemperatur von über 38 °C zweimalig innerhalb einer Stunde im Ohr gemessen bzw. über 38,3 °C einmalig im Ohr gemessen.
2.3 Blutungen
Bei einer erniedrigten Anzahl der Blutplättchen (Thrombozyten) kann die Blutstillung gestört sein. Dann kommt es zu Zahnfleischbluten und den sogenannten Petechien, kleinen punktförmigen Einblutungen in der Haut, oder Blutergüssen (Hämatomen). Diese können auch spontan, also ohne vorausgegangene Verletzung, auftreten. Bei einer gestörten Blutstillung kann bereits eine relativ leichte Blutung oder Verletzung (z.B. beim Zahnarztbesuch) bedrohlich werden. Auch bei Blutungen sollten Sie sich daher zeitnah an Ihren behandelnden Arzt wenden, damit dieser entscheiden kann, ob spezielle Maßnahmen (z.B. Thrombozytentransfusion) notwendig sind.
3 Diagnostik
Bei Vorliegen einer oder mehrerer der oben genannten Beschwerden und Symptome lässt der Hausarzt eine Untersuchung des Blutes durchführen. Wird dabei eine Unregelmäßigkeit des Blutbilds festgestellt, sollte der Patient an einen spezialisierten Facharzt mit Schwerpunkt Hämatologie und Onkologie überwiesen werden.
Dort wird eine Reihe weiterer Untersuchungen vorgenommen:
-
- mikroskopisches Differenzialblutbild
- Retikulozyten
- PNH (Paroxysmale Nächtliche Hämoglobinurie)-Diagnostik (eine relevante Zahl an PNH-Zellen ist bei bis zu 70 % der AA-Fälle nachweisbar), siehe PNH, 3 Diagnostik
-
- Hämolyse-Parameter: insbesondere LDH, Haptoglobin, Bilirubin
- Gerinnung: Quick-Wert, PTT, Fibrinogen
- Leberfunktionsparameter: AST, ALT und AP
- Nierenfunktionsparameter: Kreatinin, Harnsäure
- Blutzucker
- Gesamteiweiß, Elektrophorese, Immunglobuline
- Entzündungsparameter CRP
- Vitamin B12- und Folsäurespiegel
- Eisenstatus: Ferritin. Bei Ferritin-Werten > 1000 ng/ml weitere Abklärung von möglichen Organschäden durch eine mögliche Eisenüberladung
- Virusdiagnostik: Hepatitis A, B, C; HIV, EBV, CMV, Parvovirus B19
- Antinukleäre und Anti-DNA-Antikörper
-
- Herz- und Oberbauch-Ultraschall (Sonografie)
- Röntgenuntersuchung des Brustkorbs (Thorax)
- EKG
- HLA Typisierung des Patienten und seiner Geschwister
- Bestimmung der Telomerlängen
- Bei Verdacht auf ein „angeborenes“ Knochenmarkinsuffizienz-Syndrom weiterführende Diagnostik, z.B. Chromosomenbruchanalyse bei Verdacht auf Fanconi-Anämie, Gentests
Bestätigt sich eine erniedrigte Zahl einer oder mehrerer Blutzellreihen, ohne dass eine Ursache für einen erhöhten Verbrauch oder Abbau dieser Blutzellen bekannt ist, sollte dringend eine Untersuchung des Knochenmarks erfolgen. Auf diese Weise kann festgestellt werden, ob eine Blutbildungsstörung oder eine andere Ursache vorliegt.
Dazu wird eine Knochenmarkpunktion durchgeführt, die ambulant erfolgen kann. Dem Patienten wird dabei unter Lokalbetäubung (Lokalanästhesie) mit einer Hohlnadel (Jamshidi-Nadel) meist aus dem Beckenknochen ein Knochenzylinder entnommen (Knochenmarkbiopsie, Knochenmarkstanze). Dieser ist ca. 1,5 cm lang bei einem Durchmesser von 2-3 mm und wird mikroskopisch untersucht und beurteilt (Histologie).
Außerdem werden Blut, Knochenmark- und Fettmarkteile („Markbröckel“) gewonnen (Knochenmarkaspiration), die auf einem Objektträger ausgestrichen, getrocknet, gefärbt und ebenfalls unter dem Mikroskop in ihrer Gesamtheit bzw. in ihrer Lage zueinander beurteilt werden (zytologische Untersuchung).
Zusätzlich können an den Knochenmarkzellen genetische Untersuchungen durchgeführt werden, deren Ergebnisse z.B. die Unterscheidung von anderen Erkrankungen erleichtern können.
Da die einzelnen Laborschritte bei der Herstellung der Knochenmarkhistologie zeitintensiv sind, braucht es ca. 1–2 Wochen, bis ein vollständiges Ergebnis vorliegt. Zeigt sich eine verminderte Bildung von zwei oder drei Zellreihen (Erythrozyten, Leukozyten, Thrombozyten) entsprechend der Diagnosekriterien ohne das gleichzeitige Vorliegen von krankhaft veränderten Zellen (z.B. Leukämiezellen) und ohne dass eine Chemotherapie oder Strahlentherapie vorangegangen ist, spricht man von einer Aplastischen Anämie.
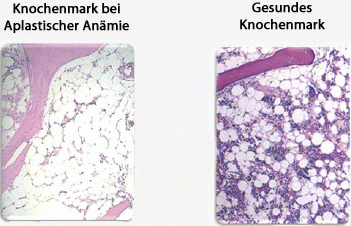
Knochenmarkbefund bei einem Patienten mit Aplastischer Anämie im Vergleich zu gesundem Knochenmark. Im erkrankten Knochenmark sind vor allem Bindegewebe und Fettzellen erkennbar. Im gesunden Mark heben sich die Blutzellen als unterschiedlich gefärbte kleine Punkte von den großen weißen Fettzellen ab.
Ziel dieser zahlreichen Untersuchungen ist es,
- andere Erkrankungen auszuschließen,
- die möglichen Ursachen abzuklären (Ätiologie),
- den Schweregrad der Aplastischen Anämie festzustellen,
- die Prognose zu ermitteln.
Bei Patienten mit sehr schwerer und schwerer Aplastischer Anämie, die unter 50 Jahre alt und bei guter körperlicher Verfassung sind, ist es sinnvoll, direkt bei Diagnosestellung eine sogenannte HLA-Typisierung des Patienten durchzuführen. Sind Geschwister vorhanden, sollten diese ebenfalls typisiert werden, um ihre Eignung für eine Stammzellspende festzustellen.
4 Verlauf
Ohne spezifische Therapie verläuft die Aplastische Anämie im Erwachsenenalter bei bis zu 70 % der Fälle tödlich.
Es besteht die Möglichkeit, dass die Aplastische Anämie in ein Myelodysplastisches Syndrom (MDS) oder eine Akute Myeloische Leukämie (AML) übergeht. Außerdem liegt bei einigen AA-Patienten eine PNH-spezifische Mutation vor, sodass auch Symptome und Therapienotwendigkeit einer Paroxysmalen Nächtlichen Hämoglobinurie (PNH) relevant werden können.
5 Therapie
5.1 Übersicht
Hämatologische Spontanheilungen (spontane Remissionen) kommen bei schwerem Knochenmarkversagen praktisch nicht vor.
Eine Notwendigkeit zur Behandlung besteht bei
- sehr schwerer (vSAA) und schwerer Aplastischer Anämie (SAA)
- nicht schwerer Aplastischer Anämie (nSAA) mit deutlicher Erniedrigung mindestens einer Zellreihe (Zytopenie), die regelmäßigen Transfusionsbedarf bedingt oder zu einer Gefährdung durch Infekte oder Blutungen führt
Während es vor einigen Jahrzehnten kaum eine Aussicht auf Heilung oder langfristige Besserung gab, gibt es heute vielversprechende Möglichkeiten. Zur Behandlung stehen vorrangig zwei Therapiemaßnahmen zur Verfügung: die sogenannte immunsuppressive Therapie (IST) und die Stammzelltransplantation (SZT) bzw. Knochenmarktransplantation (KMT). Darüber hinaus gibt es für bestimmte Untergruppen von Patienten spezielle Therapien. Welche davon für einen bestimmten Patienten infrage kommt, ist abhängig von der Schwere der Erkrankung, dem Alter und möglichen Begleiterkrankungen des Patienten sowie vom Grad der sogenannten HLA-Übereinstimmung (HLA-Kompatibilität) mit einem verwandten oder nicht verwandten möglichen Knochenmarkspender.
Besteht eine Therapieindikation, sollte die Behandlung möglichst schnell begonnen werden, um das Fortschreiten der Erkrankung und deren mögliche Komplikationen (z.B. ausgeprägte Anämie, Infektionen, Blutungen und Gerinnungsstörungen) zu vermeiden. Daher ist eine frühzeitige Therapieplanung in Zusammenarbeit mit einem spezialisierten Zentrum wichtig.
Der Therapieablauf für Patienten mit sehr schwerer und schwerer Aplastischer Anämie wird in der folgenden Abbildung dargestellt.
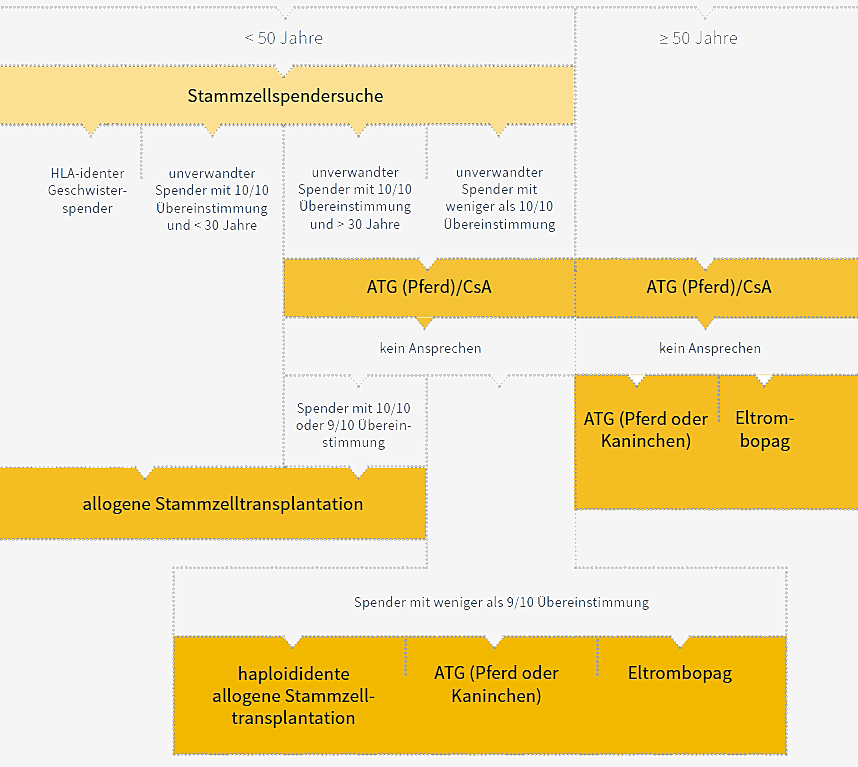
Deutlich vereinfachte Darstellung des Therapiealgorithmus. Der vollständige Algorithmus ist abgebildet in den Leitlinien der DGHO auf Onkopedia
5.2 Immunsuppressive Therapie (IST)
Antithymozytenglobulin (ATG) und Ciclosporin (CsA)
Da sich bei der erworbenen Aplastischen Anämie das körpereigene Immunsystem gegen das eigene Knochenmark wendet, ist oftmals eine immunsuppressive Therapie angezeigt, vor allem bei:
- Patienten mit vSAA oder SAA > 50 Jahre
- Patienten ohne HLA-identen (Geschwister-)Spender
- Patienten mit vSAA oder SAA < 50 Jahre in schlechtem körperlichen Zustand
- Patienten mit nSAA/MAA mit Gefährdung durch schwere Zytopenie in mindestens einer Zellreihe
Bei der immunsuppressiven Therapie handelt es sich zumeist um eine Kombination aus den Medikamenten Antithymozytenglobulin und Ciclosporin. Dadurch kann sich das Knochenmark wieder erholen. Im Verlauf der Therapie kommt es zunächst meist kurzfristig zu einer Verschlechterung der Blutbildsituation, ehe eine Besserung eintritt.
ATG ist ein Antikörper, der die überaktiven, knochenmarkschädigenden T-Lymphozyten zerstört. Üblicherweise wird ATG für 4–5 Tage als Infusion in eine große Vene über einen zentralen Venenkatheter (ZVK) gegeben. Während der ATG-Therapie sollte der Thrombozytenwert ggf. mittels Thrombozytentransfusion auf 30 G/l angehoben bzw. dort gehalten werden, da es unter der Therapie zu einem raschen Thrombozytenabfall kommen kann. Für eine ATG-Therapie muss man mit einem stationären Aufenthalt von ca. 1-2 Wochen rechnen. Nebenwirkungen der ATG-Therapie können allergische Reaktionen wie Hautausschlag und Fieber sein. Zur Unterdrückung von akuten Nebenwirkungen des ATG wird zusätzlich für eine kurze Zeit ein Kortisonpräparat, z.B. Prednison oder Prednisolon, verabreicht. Außerdem besteht ein erhöhtes Risiko für bestimmte Erreger, sodass diesbezügliche Vorsichtsmaßnahmen getroffen werden müssen.
Ein weiterer wesentlicher Faktor für das Therapieansprechen der Erkrankung ist das Ciclosporin, das die Ausschüttung immunstimulierender Stoffe hemmt. Bei CsA werden regelmäßige Laborkontrollen durchgeführt, um ggf. durch eine Dosisanpassung die optimale Wirkung zu erzielen. Dabei wird ein Talspiegel von 170–225 ng/ml im Blut angestrebt. Für einen stabilen Wirkspiegel sollte die Medikamenteneinnahme sehr regelmäßig in einem festen zeitlichen Abstand von 12 Stunden erfolgen.
Mögliche Nebenwirkungen der CsA-Therapie sind Infektionen, Verschlechterung der Nierenfunktion, Erhöhung des Blutdrucks, Zahnfleischwucherung (Gingivahyperplasie), Vermehrung des Haarwuchses, Muskelkrämpfe, Sensibilitätsstörungen oder Zittern (Tremor). Die Nebenwirkungen sind abhängig von der Dosis und verschwinden üblicherweise bei Beendigung der CsA-Therapie wieder.
CsA wird als Kapsel oder Saft für mindestens 12 Monate eingenommen. Bei einem sehr guten und stabilen Therapieansprechen ist es für das Absetzen wichtig, die CsA-Dosis sehr langsam und schrittweise auszuschleichen, um einen Krankheitsrückfall zu vermeiden. Bei einem Teil der Patienten muss CsA jedoch länger oder dauerhaft gegeben werden, um den Therapieerfolg aufrechtzuerhalten.
Durch die intensivierte immunsuppressive Therapie kann bei etwa 50–75 % der Patienten eine Heilung (komplette Remission, CR) oder zumindest eine deutliche Besserung (partielle Remission, PR) erzielt werden, bei der Transfusionsunabhängigkeit und eine deutliche Reduktion des Infektions- und Blutungsrisikos besteht. Es dauert etwa 2-4 Monate, bei manchen Patienten auch 6 Monate, bis eine Besserung der Blutwerte eintritt. Das Ziel ist die Beendigung der klinischen Beschwerden und Risiken. Dafür ist eine vollständige Normalisierung der Blutwerte, die häufig nicht erreicht wird, nicht nötig.
Bei fehlendem Ansprechen kann eine Wiederholung der immunsuppressiven Therapie nach 4-6 Monaten erwogen werden.
Bei einem Rückfall (Rezidiv) ist eine Wiederholung der immunsuppressiven Therapie ebenfalls möglich, da die Chance auf ein erneutes Ansprechen bei 30-60 % liegt.
Begleitend zu einer spezifischen Therapie sollte jeder Patient eine sogenannte unterstützende (supportive) Therapie erhalten.
Alemtuzumab
Es gibt auch andere Medikamente, die über den gleichen Mechanismus der Immunsuppression wirken. Dazu gehört z.B. Alemtuzumab, ein Antikörper, der gegen T-Lymphozyten wirkt. Dieses Medikament wird bei der Chronischen Lymphatischen Leukämie (CLL) oder der Multiplen Sklerose (MS) eingesetzt, konnte aber auch bei der Aplastischen Anämie in Studien gerade bei älteren Patienten gute Ansprechraten zeigen. Ein Vorteil dieses Medikaments ist, dass es nur unter die Haut gespritzt wird, dafür also kein stationärer Aufenthalt notwendig ist. Wenn der Patient früher eine Infektion mit dem Cytomegalie-Virus (CMV) hatte, sollte dieser Blutwert regelmäßig kontrolliert werden, da diese Virusinfektion unter Therapie wieder auftreten kann.
Patienten, bei denen andere Therapien nicht wirkten, zeigten unter Alemtuzumab Ansprechraten von 37-48 %.
5.3 Allogene Transplantation
Bei Patienten bis zu einem Alter von ca. 50 Jahren (wichtig ist der Allgemeinzustand, „das biologische Alter“) mit einer schweren oder sehr schweren Aplastischen Anämie (SAA oder vSAA) und Verfügbarkeit eines Geschwisterspenders, der in den Gewebsverträglichkeitsstrukturen (HLA) mit dem Patienten vollständig übereinstimmt (HLA-ident), ist die bevorzugte Behandlung (Erstlinientherapie) eine allogene Transplantation.
Patienten, die jünger als 30 Jahre alt sind, können auch die Stammzellen eines nicht verwandten HLA identen Spenders (Fremdspenders) erhalten, wenn sie keinen HLA-identen Familienspender besitzen. Wichtig ist, dass eine sogenannte Feintypisierung (mindestens 10 HLA-Antigene) vorgenommen wird und Spender und Empfänger in dieser komplett identisch sind.
In den letzten Jahren konnte die Komplikationsrate bei der HLA-identen Fremdspendertransplantation deutlich reduziert werden. Die HLA-idente Fremdspendertransplantation wird daher auch zunehmend bei Patienten angewandt, die jünger als 50 Jahre sind und nicht auf eine immunsuppressive Behandlung angesprochen haben.
Das Ziel der allogenen Transplantation ist es, das nicht funktionsfähige Knochenmark des Patienten durch gesunde Stammzellen eines Spenders zu ersetzen. Hierfür wird zuerst durch verschiedene Maßnahmen (Chemotherapie, Antikörpertherapie, Bestrahlung) das Knochenmark des Patienten zerstört. Diese sogenannte Konditionierung wird in den Tagen direkt vor der Transplantation durchgeführt.
Parallel hierzu erfolgt die Sammlung neuer, gesunder Stammzellen bei einem gesunden, verwandten oder nicht verwandten freiwilligen Spender.
Die Stammzellen können mittels Knochenmarkbiopsie unter Narkose direkt aus dem Knochenmark gewonnen werden. Durch die Einstiche in den Beckenkamm zur Entnahme des Knochenmarks kann es zu Blutergüssen und Schmerzen kommen, die mehrere Tage anhalten. Zusätzlich besteht das allgemeine Narkoserisiko.
Alternativ dazu wird dem Spender an mehreren Tagen ein Medikament gespritzt, das die Granulozytenbildung stimuliert (G-CSF). Die dadurch vermehrt erzeugten Blutstammzellen wandern aus dem Knochenmark ins Blut. Diese sogenannten peripheren Blutstammzellen (PBSZ) werden dann wie bei einer Blutplasmaspende mit einem speziellen Gerät entnommen (Apherese). Bei dem Verfahren kann es zu grippeähnlichen Beschwerden und Schmerzen kommen.
Werden die Stammzellen direkt aus dem Knochenmark gewonnen, spricht man von einer Knochenmarktransplantation (KMT), erfolgt die Stammzellgewinnung mittels Apherese, nennt man es Stammzelltransplantation (SZT).
Studien deuten darauf hin, dass eine Therapie der Aplastischen Anämie mit Stammzellen aus dem peripheren Blut mit vermehrten Komplikationen wie z.B. akuten oder chronischen Abstoßungsreaktionen einhergehen kann. Wenn möglich, sollten daher Stammzellen verwendet werden, die direkt aus dem Knochenmark gewonnen wurden.
Unabhängig davon, wie die Stammzellen gewonnen wurden, werden sie gereinigt und auf Infektionserreger untersucht. Anschließend erhält der Patient die gespendeten Stammzellen. Die Transplantation selbst verläuft wie eine Bluttransfusion. Wenn alles gut geht, „wachsen“ die Spenderstammzellen an und führen zu einer normalen Knochenmarkfunktion und Blutbildung. Für eine Transplantation ist ein stationärer Aufenthalt von mindestens 4 Wochen erforderlich.
Während der Transplantation erhält der Patient Medikamente zur Vorbeugung (Prophylaxe) von Infektionen durch Bakterien und Pilze. Außerdem werden ein Kortisonpräparat, z.B. Prednisolon, und Ciclosporin (CsA) zur Beeinflussung des Immunsystems über mehrere Monate gegeben.
Mögliche Komplikationen durch die Transplantation sind:
- Toxische Nebenwirkungen während der Konditionierungstherapie
- Infektionen
- Graft versus Host-Disease (GvHD)
- Transplantatabstoßung
Graft versus Host-Disease (GvHD): Hierbei reagiert das gespendete Immunsystem gegen die körpereigenen Zellen. Dies kann kurzzeitig (akut) oder später und langanhaltend (chronisch) erfolgen, sodass unter Umständen eine dauerhafte Unterdrückung des Immunsystems (immunsuppressive Therapie) notwendig werden kann.
5.4 Weitere Therapieoptionen
Danazol
Bei manchen Patienten mit Aplastischer Anämie liegt eine seltene angeborene Störung vor, bei der die Enden der Chromosomen (Telomere) verkürzt sind, was als Telomeropathie bezeichnet wird. Die Verkürzung der Telomere führt zu einer Zellteilungsstörung und damit zu einer verringerten Bildung von Blutzellen im Knochenmark.
Für Danazol, eine synthetische Variante des männlichen Sexualhormons Testosteron, konnte gezeigt werden, dass es eine Verlängerung der Telomere bewirken kann.Dies kann zu einer Verbesserung der Symptome bis hin zu einer Normalisierung der Blutbildung führen.
Eltrombopag (Revolade®)
Seit 2015 ist Eltrombopag bei erwachsenen Patienten mit erworbener schwerer Aplastischer Anämie (SAA) zuglassen, wenn sie
- entweder auf eine vorangegangene immunsuppressive Therapie nicht angesprochen haben oder
- stark vorbehandelt und
- für eine Knochenmark- bzw. Stammzelltransplantation nicht geeignet sind.
Aufgrund sehr guter Ansprechraten in klinischen Studien wurde Eltrombopag in den USA bereits in Kombination mit hATG und CsA für die Erstlinienbehandlung der Aplastischen Anämie zugelassen. Eine Zulassung für die Erstbehandlung liegt in der EU nicht vor. Aufgrund der nachgewiesenen Wirksamkeit empfiehlt die Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie jedoch die Verwendung von Eltrombopag bei der Erstlinientherapie der erworbenen SAA/vSAA im Rahmen ihrer Leitlinien.
Eltrombopag wirkt auf die Steuerung der Blutstammzell- und Thrombozytenbildung ein. Das Medikament aktiviert das Thrombopoetin, das die Bildung der Thrombozyten und des Bluts (Hämatopoese) steuert. Die Wirkdosis bei der SAA beträgt
150 mg/Tag. Es konnte gezeigt werden, dass durch den Einsatz von Eltrombopag bei den meisten Patienten sowohl eine Verbesserung der Thrombozyten- als auch der Erythrozyten- und Neutrophilenwerte zu verzeichnen ist. Ebenso kam es unter Eltrombopag zu einer Verbesserung bzw. Normalisierung der Knochenmarkzellularität. Bei Patienten mit zuvor regelmäßigem Transfusionsbedarf verlängerte sich die Anzahl der Tage bis zur nächsten Transfusion bzw. sie wurden transfusionsunabhängig.
Sonstiges
Therapien ohne nachgewiesene Wirksamkeit, z.B. Steroid-Monotherapie oder Monotherapie mit hämatopoetischen Wachstumsfaktoren, sollten unterlassen werden, da sie nur Zeitverlust bedeuten und die Ausgangssituation des Patienten im Hinblick auf eine der bewährten Therapiemöglichkeiten wesentlich verschlechtern können.
5.5 Unterstützende (Supportive) Therapie
Die Vorbeugung und Behandlung von Infektionen, die Vermeidung von Blutungen, die individuell angepasste Transfusionsstrategie und die Behandlung einer Eisenüberladung sind bei der unterstützenden Therapie von besonderer Bedeutung. Diese Maßnahmen konnten in den vergangenen Jahren verbessert werden und tragen zu einer verbesserten Überlebenswahrscheinlichkeit bei, auch wenn durch die Behandlung kein komplettes Ansprechen erreicht wird.
Infektionen
Bei fieberhaften Infektionen sollte möglichst unverzüglich ein Arzt aufgesucht werden, um eine Diagnose zu stellen und eine Therapie einzuleiten.
In bestimmten Fällen kann der vorbeugende Einsatz von Antibiotika gegen Bakterien und von Antimykotika gegen Pilze sinnvoll sein.
- Bei Patienten mit schwerer Neutropenie (Granulozyten-/Neutrophilenzahl < 0,5 G/l)
- Bei Patienten unter Antithymozytenglobulin-Therapie (ATG) oder Alemtuzumab sollte eine zusätzliche Vorbeugung gegen weitere Erreger (z.B. Pneumocystis jirovecii, Cytomegalie-Virus) durchgeführt werden.
Außerdem sollten bei erniedrigter Granulozyten-/Neutrophilenzahl (< 0,5 G/l) verschiedene Verhaltensmaßnahmen beachtet werden:
- Kontakt mit Menschen meiden, die an Infektionen erkrankt sind
- engen körperlichen Kontakt zu Tieren meiden
- große Menschenansammlungen meiden, insbesondere in den Wintermonaten
- übliche Hygienemaßnahmen durchführen, z.B. Händewaschen, Mundpflege, bei rohen Lebensmitteln auf Frische und Reinigung achten
- engen Kontakt mit Pilzsporen meiden, v.a. Gartenarbeit, keine Biotonne reinigen oder Kompost umsetzen
In sehr seltenen Fällen, z.B. bei schweren Infektionen, kann der Einsatz der hämatopoetischen Wachstumsfaktoren G-CSF bzw. GM-CSF zur Anregung der körpereigenen Immunabwehr oder die Transfusion von Konzentraten weißer Blutkörperchen (Granulozytenkonzentrate) erwogen werden.
Weitergehende Informationen finden Sie in der Broschüre „Infektionen? Nein, danke!” von M. Exner und A. Simon der Stiftung Deutsche Leukämie- & Lymphom-Hilfe.
Blutungen
Durch die reduzierte Zahl an Blutplättchen können schwere, auch lebensbedrohliche Blutungen bei Aplastische Anämie-Patienten auftreten. Es kann sowohl zu einer verstärkten oder verlängerten Blutung bei Verletzungen oder Operationen kommen als auch zu plötzlichen Blutungen ohne erkennbare Ursache.
Umso wichtiger ist es, die Funktion der vorhandenen Thrombozyten nicht durch Medikamente einzuschränken. Solche Medikamente sind z.B. Thrombozytenaggregationshemmer, da diese das Zusammenklumpen der Blutplättchen verhindern und daher die Blutstillung stören.
Umso wichtiger ist es, die Funktion der vorhandenen Thrombozyten nicht durch Medikamente einzuschränken. Solche Medikamente sind z.B. Thrombozytenaggregationshemmer, da diese das Zusammenklumpen der Blutplättchen verhindern und daher die Blutstillung stören. Der Einsatz von Thrombozytenaggregationshemmern wie z.B. Acetylsalicylsäure (ASS) ist daher insbesondere bei sehr niedriger Thrombozytenzahl als äußerst kritisch zu bewerten und sollte sorgfältig abgewogen werden.
Bei Frauen mit starken Auswirkungen ihrer Menstruationsblutungen kann zudem zur Vermeidung von übermäßigen Blutverlusten bei bestehendem Thrombozytenmangel die Menstruationsblutung mittels einer Hormon-Therapie, z.B. durchgehende Gabe der Pille oder 3-Monats-Spritze, vorübergehend ausgeschaltet werden.
Das wesentliche Mittel zur Vermeidung von Blutungen und Therapie bestehender Blutungen ist die Transfusion von Thrombozytenkonzentraten.
Blutübertragungen (Transfusionen)
Transfusionen sind bei vielen Patienten zur Sicherung einer ausreichenden körperlichen Belastbarkeit und Lebensqualität sowie zur Vermeidung von Blutungskomplikationen erforderlich. Sie können bei entsprechenden Symptomen (Anämie, Blutungen) vorübergehend die fehlenden Blutzellen ersetzen. Dabei wird nicht das gesamte Blut übertragen (transfundiert), sondern nur jeweils die Zellart, die benötigt wird (rote Blutkörperchen oder Blutplättchen).
Für die Herstellung eines Konzentrats wird nach einer Blutspende das Blut zunächst untersucht, um übertragbare Infektionen auszuschließen, dann werden die weißen Blutkörperchen entfernt, und im Anschluss werden die verschiedenen Blutbestandteile aufgetrennt und konzentriert. Familienmitglieder sind als Blutspender nicht zulässig, da gerade in diesen sogenannten gerichteten Spenden besondere Risiken liegen.
Übertragung von roten Blutkörperchen/Erythrozytenkonzentraten (EK-Transfusion)
Um eine gute Verträglichkeit zu gewährleisten, wird das verwendete Präparat nicht nur nach der Blutgruppe (A, B, AB, 0) und dem Rhesusfaktor ausgesucht, sondern jedes einzelne Präparat wird individuell für den jeweiligen Patienten ausgetestet. Dafür wird bei einer Verträglichkeitsprobe, der sogenannten Kreuzprobe, Blut des Patienten mit Blut aus dem Erythrozytenkonzentrat (EK) gemischt und untersucht. Insbesondere wenn Antikörper vorliegen, kann es länger dauern, ein geeignetes Erythrozytenkonzentrat zu finden. Ist ein Patient Cytomegalie-Virus-(CMV-)negativ und besteht die Möglichkeit einer späteren Knochenmark- oder Stammzelltransplantation, sollten CMV-negative Konzentrate gegeben werden.
Generell sollten Erythrozytenkonzentrate zurückhaltend eingesetzt werden, da sie zu einer Überladung des Körpers mit Eisen führen können. Indikationen für eine Transfusion sind
- ausgeprägte Leistungsminderung verbunden mit Müdigkeit oder im Rahmen einer Kurzatmigkeit, z.B. unter körperlicher Belastung, und in Abhängigkeit von den jeweiligen Begleiterkrankungen, z.B. Herzinsuffizienz
- ein sehr niedriger Hämoglobinwert (< 8 g/dl)
Generell gilt bezüglich der Transfusionstherapie:
So viel wie nötig, so wenig wie möglich!
Die Transfusionsindikation muss daher anhand folgender Punkte individuell für jeden Patienten abgewogen werden:
- klinische Symptome (z. B. Herzfrequenz, Atemfrequenz, Kurzatmigkeit, Blutungszeichen, Fieber, Infektion…)
- subjektive Beschwerden (z. B. deutliche Leistungsminderung, Müdigkeit, Schwäche, Kopfschmerzen, Pochen in
den Ohren..) - individuelle Begleiterkrankungen/Vorgeschichte (z.B. Herzinsuffizienz, Blutung)
- Medikamente/Therapien (z.B. ATG-Therapie, operativer Eingriff)
- Möglichkeit zur Überwachung und kurzfristigen Transfusion (ambulant/stationär)
Die zurückhaltende Transfusionsstrategie gilt vor allem für Patienten, bei denen eine allogene Stammzelltransplantation geplant ist. Bei geplanter Stammzelltransplantation sollen keinesfalls Transfusionen von Blutprodukten von Angehörigen erfolgen.
Übertragung von Blutplättchen/Thrmobozytenkonzentraten (TK-Transfusion)
Bei Blutungen können Konzentrate von Blutplättchen (Thrombozytenkonzentrate, TK) übertragen werden, um Komplikationen zu vermeiden. Da Blutungen bei niedrigen Thrombozytenzahlen lebensbedrohliche akute Notfälle sein können, muss in diesen Fällen sofort gehandelt werden. Sind die Thrombozytenzahlen sehr niedrig, können Thrombozytenkonzentrate auch vorbeugend gegeben werden. Die Lebensspanne von Blutplättchen beträgt nur wenige Tage. Bei einer sehr geringen bis fehlenden Produktion von Thrombozyten im Knochenmark kann die Gabe von Thrombozytenkonzentraten daher mehrmals pro Woche nötig sein.
Thrombozyten tragen Gewebemerkmale (HLA-Marker), die für jede Person unterschiedlich sind. Bei einem Teil der Patienten treten Antikörper gegen diese HLA-Marker auf. Dies kann spontan, im Rahmen von Erkrankungen oder nach Schwangerschaften geschehen. Liegen solche HLA-Antikörper vor, werden die transfundierten Blutplättchen sofort zerstört, es kommt zu keinem ausreichenden Thrombozytenanstieg nach einer Transfusion eines Thrombozytenkonzentrats. Für diese Patienten müssen spezielle HLA-kompatible Thrombozytenkonzentrate hergestellt werden von Spendern mit passenden HLA-Merkmalen.
Die Indikation für eine Transfusion von Thrombozytenkonzentraten besteht:
- bei Thrombozyten-Werten < 20 G/l und Fieber >38° C, Infektionen, Blutungszeichen oder einer Vorgeschichte von schweren Blutungen (WHO Grad 3 oder 4) sowie bei Alloimmunisierung (HLA-Antikörper, siehe oben)
- wenn keine Risiken vorliegen, die die Blutungsgefahr erhöhen (z.B. Fieber, Infektionen, schwere Blutung in der Vorgeschichte, Alloimmunisierung) kann auch erst bei Thrombozytenwerten < 5 G/l transfundiert werden. Voraussetzung dafür sind regelmäßige und engmaschige Kontrollen (z. B. mindestens einmal pro Woche), das Fehlen von Blutungszeichen (z.B. Petechien), und die Möglichkeit einer raschen Transfusion bei Blutungszeichen.
- „Patienten-individueller Grenzwert“: Viele Patienten haben einen stabilen Grenzwert, bei dessen Unterschreiten stärkere Blutungszeichen auftreten.
- Vor und während der ATG-Therapie soll der Thrombozytenwert auf 50 G/l angehoben werden, da es unter ATG-Infusion zu einem raschen Thrombozytenabfall kommen kann.
- Vor Operationen oder Eingriffen sollte der behandelnde Arzt unbedingt vorab über die Aplastische Anämie informiert werden und am besten ein aktuelles Blutbild vorgelegt bekommen!!
Eisenüberladung
Mit jedem Erythrozytenkonzentrat wirdmehr als das 100fache der Menge an Eisen aufgenommen, die täglich mit der Nahrung zugeführt wird. Da der menschliche Körper Eisen nicht aktiv ausscheiden kann, lagert es sich in verschiedenen Organen, v.a. Leber, Herz, Niere, Knochenmark ab (siehe Tabelle 2) und kann sie schädigen. Patienten mit Aplastischer Anämie oder Myelodysplastischem Syndrom (MDS) haben bereits ohne Transfusionen ein Risiko für eine Eisenüberladung, da durch die schlechte Knochenmarkfunktion die Neubildung von Erythrozyten vermindert ist und das Eisen nicht vollständig für die Bildung neuer roter Blutkörperchen verwendet werden kann.
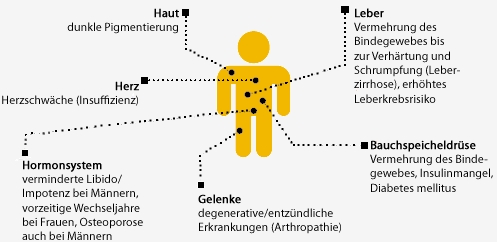
Komplikationen einer Eisenüberladung
In der Regel sind in den ersten Monaten nach Diagnose noch keine Speichereisen (Ferritin)- bzw. Lebereisenwerte erreicht, die eine sofortige Therapie zur Bindung und Ausscheidung des überschüssigen Eisens (Chelattherapie) erfordern. Man sollte daher bis mindestens 6 Monate nach Einleitung der Immunsuppression abwarten. Bei andauernder regelmäßiger Transfusionsbedürftigkeit ist bei Serumferritin-Spiegeln über 1.000 µg/l eine Chelattherapie angezeigt. Dies gilt insbesondere auch für Transplantationskandidaten, da eine Eisenüberladung mit höherer transplantationsverbundener Sterblichkeit (Mortalität) und höherer Krankheitshäufigkeit (Morbidität) einhergeht.
Die heutzutage eingesetzten Medikamente zur Behandlung einer Eisenüberladung sind im Allgemeinen gut verträglich. Zu den wesentlichen Nebenwirkungen gehören Übelkeit, Durchfälle und Nierenfunktionsstörungen, die sich jedoch nach Absetzen zurückbilden.
Wenn das Serumferritin unter 1.000 µg/l ist, kann bei transfusionsbedingter Eisenüberladung eine Unterbrechung der Behandlung in Abhängigkeit vom individuellen Transfusionsbedarf in Erwägung gezogen werden. Dies sollte jedoch immer in Rücksprache mit dem behandelnden Arzt erfolgen.
Bei Transfusionsfreiheit und ausreichend hohem Hämoglobinwert ist die Aderlasstherapie (Menge individuell, z. B. mit geringen Mengen wie 100 ml pro Entnahme) als nebenwirkungsarme effektive Therapiemöglichkeit der Eisenüberladung zu empfehlen.
Generell ist die Eisenüberladung regelmäßig zu überprüfen und die Therapie anzupassen.
Weitergehende Informationen finden Sie in der Broschüre „Transfusionsbedingte Eisenüberladung” der Stiftung Deutsche Leukämie- & Lymphom-Hilfe.
Aktivitäten
Bei Patienten mit Aplastischer Anämie sind körperliche Aktivitäten und Bewegung in Abhängigkeit von den Blutwerten und dem Befinden sinnvoll. Es sollte jedoch darauf geachtet werden, dass keine Überforderung auftritt. Daher ist es ratsam, Sport unter Pulskontrolle zu betreiben. Dies ist insbesondere bei Vorliegen einer Anämie wichtig, da bei einer Verminderung der Erythrozyten der Körper häufig versucht, diesen Mangel durch einen gesteigerten Herzschlag auszugleichen, was zu einer zu starken Belastung des Herzens führen kann. Bei Thrombozytopenie sollte unbedingt auf verletzungsträchtige Sportarten, z.B. Kampfsport oder Klettern, verzichtet werden.
Rehabilitation
Sollte aufgrund der Aplastischen Anämie eine Teilnahme am „normalen“ Leben nicht mehr wie gewohnt möglich sein, können eine Rehabilitationsmaßnahme, ambulante Physiotherapie bzw. Krankengymnastik oder eine psychologische oder psychotherapeutische Betreuung sinnvoll sein. Diese Maßnahmen sollten individuell auf den Patienten abgestimmt werden.
Sind intensive Therapiemaßnahmen geplant, ist es sinnvoll, die Rehabilitationsmaßnahmen erst nach diesen Therapien durchzuführen. Krankengymnastik bzw. Physiotherapie oder eine psychologische beziehungsweise psychotherapeutische Betreuung sind auch therapiebegleitend hilfreich.
Bei der Wiedereingliederung eines an Aplastischer Anämie erkrankten Kindes nach Abschluss der immunsuppressiven Therapie bzw. Transplantation kann wegen der hohen psychosozialen Belastung der Familien eine familienorientierte Maßnahme in einer pädiatrisch-onkologischen Nachsorgeeinrichtung sinnvoll sein.
6 Prognose
Je höher die Granulozytenzahl und je geringer das Alter des Patienten zum Zeitpunkt der Diagnose ist, desto besser ist die Prognose.
Bei den im Folgenden genannten Daten zum Überleben nach verschiedenen Therapien handelt es sich um statistische Daten. Dies bedeutet, dass sie nicht automatisch auf den einzelnen Patienten zu übertragen sind. Diese Auflistung soll nur einen Überblick darüber geben, wie sich die Möglichkeiten und das Überleben in den letzten Jahren verbessert haben. Es gibt häufig Untergruppen, die hier nicht berücksichtigt sind. Bei allen spezifischen Therapieformen sind die Ergebnisse für Patienten unter 20 Jahren deutlich besser als für Patienten über 20 Jahre. Das Gleiche gilt für Patienten unter 40/50 Jahren gegenüber Patienten über 40/50 Jahre. Bei den Stammzelltransplantationen sind die Ergebnisse erheblich besser, wenn der Spender Stammzellen direkt aus dem Knochenmark und nicht die aus dem Blut gewonnenen peripheren Stammzellen zur Verfügung stellt.
Veröffentlichten Daten zufolge beträgt das Gesamtüberleben bei der SAA/vSAA nach 3-6 Jahren und getrennt nach den verschiedenen spezifischen Therapieformen:
- nach allogener SZT von HLA-identem Familienspender 75–90 %
- nach allogener SZT von HLA-identen nicht verwandten Spendern 65–73 %
- nach ATG/CsA-Therapie 76–96 %
7 Register
Patienten mit dem Nachweis eines PNH-Klons können in das internationale PNH-Register (PNH Registry) über das Universitätsklinikum Essen aufgenommen werden, damit weitere Erkenntnisse über diese Gruppe von AA-Patienten gewonnen werden können.
Bei Interesse oder Rückfragen wenden Sie sich bitte per E-Mail an Prof. Dr. med. Alexander Röth (alexander.roeth(at)uk-essen.de).
Patienten mit einer Telomeropathie können in das AA-BMF-Register im Universitätsklinikum Aachen aufgenommen werden, damit Erkenntnisse über diese Untergruppe von AA-Patienten gewonnen werden können.
Bei Interesse oder Rückfragen wenden Sie sich bitte per E-Mail an Prof. Dr. med. Tim H. Brümmendorf (tbruemmendorf(at)ukaachen.de).